

Department of Health Informatics
- M.S. Health Informatics
- M.S. Clinical Research Management
- MS in Health Information Management
- Doctor of Health Informatics
- B.S. Health Information Management
- Health Information Management
- Healthcare Informatics
Health informatics is the most promising biomedical science and information technology frontier of the 21st century. The health informatics field is revolutionizing the health care and pharmaceutical industries in countless ways, using information and technology to improve health care and advance biomedical research.
To address a critical need for health informatics training, the Rutgers SHP Department of Health Informatics offers a complete career ladder in the fields of health data analytics, health information management, and clinical research management. Programs are available as completely online, completely on-campus, or a combination of both.
Our health informatics graduates find employment immediately in health care/pharmaceutical sectors as clinical data analysts, regulatory affairs professionals, clinical research associates, drug safety officers, and medical affairs professionals, as well as faculty or researchers at universities and research facilities.
Department Chair: Shankar Srinivasan, PhD
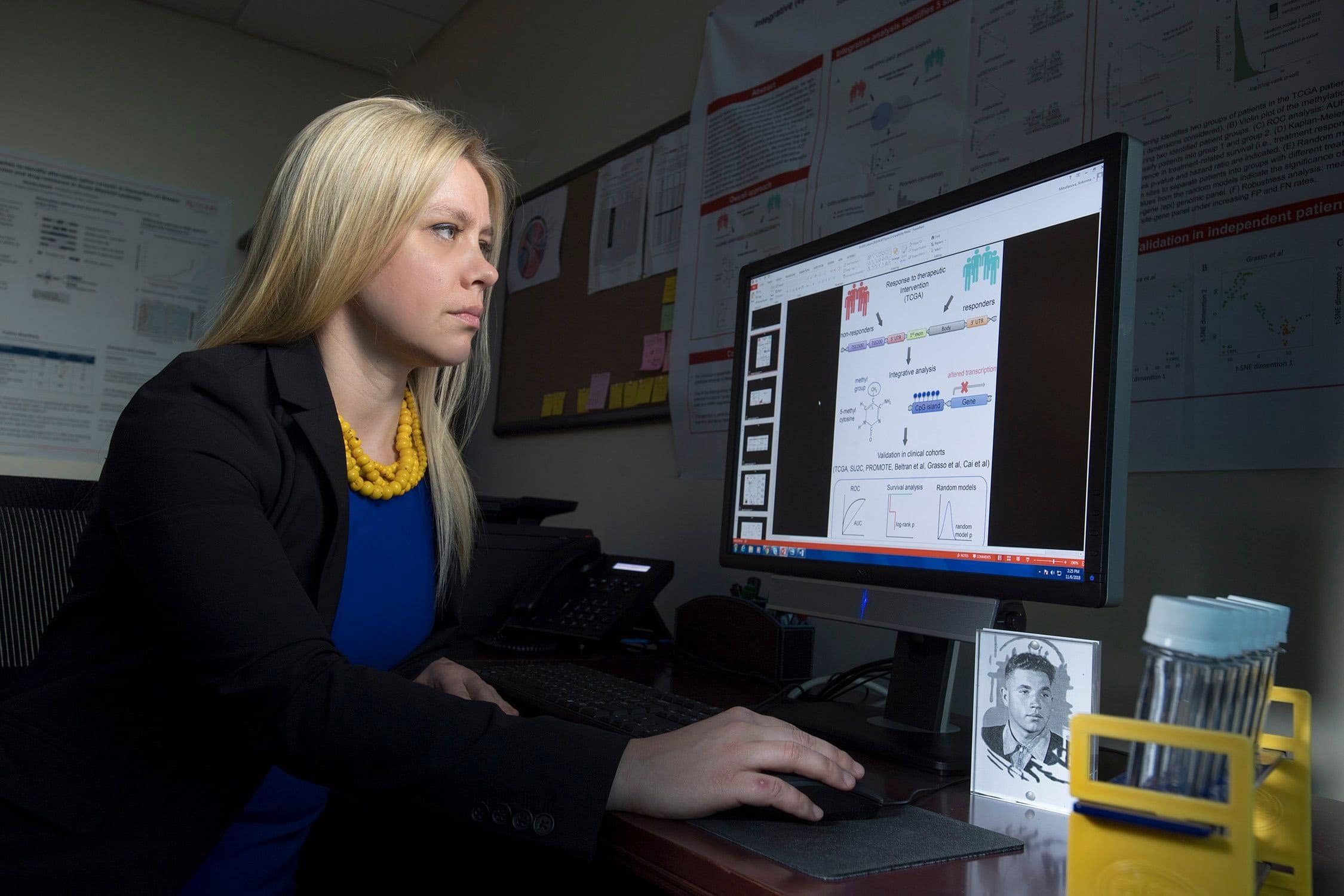
Antonina Mitrafanova, pioneer in health informatics
Mitrafanova has developed an algorithm that predicts how prostate cancer patients will respond to androgen deprivation therapy (ADT). A patent is pending.
Read her inspiring story in Rutgers Today.









Rutgers is an equal access/equal opportunity institution. Individuals with disabilities are encouraged to direct suggestions, comments, or complaints concerning any accessibility issues with Rutgers web sites to: accessibility@rutgers.edu or complete the Report Accessibility Barrier or Provide Feedback Form.